Industries
March 2023
Standards are documents that summarize specifications, procedures, and guidelines to ensure that a product is safe, consistent, and reliable. For medical electrical equipment, electrical safety standards are of paramount importance. However, even though they are primarily intended to provide guidance and assistance, they are often perceived as a challenge. As a development partner, Helbling has extensive experience in working with the IEC 60601 family of standards, covering the Basic Safety and Essential Performance of medical electrical equipment. Below, Helbling’s experts share five top tips to help with meeting requirements without getting lost in the effort.
Tip 1: Compliance is not possible if the standard is only considered at the end of development.
It is often found that companies plan compliance with norms as part of the verification process and that no activities are started before that stage.
This is a fundamental mistake: fulfilling the IEC 60601 standard is a complex and multifaceted task; it is not just a list of tests to be added to the verification plan. The IEC 60601 standard family provides guidance in designing medical electrical equipment properly. If this is not considered early enough, there is a possibility of missing a substantial set of design input requirements that should not be neglected or cannot be fixed easily retrospectively.
The earlier this is considered during development, the greater the chances that it will work out with fewer development loops, shortening time to market.
Tip 2: Usability, documentation, and risk management is key.
Besides testing, compliance with the norm also includes reviewing the main design documents. Usability and risk management files are particularly important here.
Early risk analysis will help to define the device’s Essential Performance. This relates to clinical functions, other than Basic Safety, the loss or degradation of which results in unacceptable risk.
At the start of development, decisions are made on defining the product vision and key clinical claims, who will be using the product (both patients and clinicians), how it will look, where it will be used, etc.
Accordingly, an important task is to define the types of users who may touch or use the product. If a medical device is only used by well-trained professionals in a controlled environment, certain tests do not need to be performed or the risks can be mitigated with additional warnings in the instructions for use. If the device is used by a patient alone at home, risk mitigations are generally more conservative because the user’s age group, general knowledge, and degree of impairment must also be considered (a collateral standard dedicated to home healthcare equipment addresses this point). In addition, the environment in which the device is used needs to be defined in the risk management file to determine the limits against which the device is tested (for instance: professional healthcare facility, home healthcare, special environment). As such, the electromagnetic emission and interference limits to be considered for a hospital monitor used in a controlled environment are very different from the limits to be considered for a medical device for home use.
Tip 3: The standard should be understood correctly.
The latest valid version of the standard should be used. The standard amendments can sometimes affect key aspects of the design. For example, new digital wireless technologies were not considered in former versions of the norm. Applicable collateral standards specific to the medical electrical equipment should be considered as they may overrule the general standard.
Standards are not free from misinterpretation. This can lead to missing things that are important or overdesigning to comply with sections that do not even apply. For instance, Helbling has found on occasion that companies have designed the medical device housing with the requirements of a fire enclosure when that has not applied given the low power level.
The standards offer some freedom on how to be compliant. As an example, sometimes it is possible either to add extra shielding to a device to protect it against electromagnetic interference or simply to write in the instructions for use that the device should be moved away from sources of interference. A risk analysis must be performed with documentation of the mitigation and the patient must be informed.
If in doubt, it is worth investing in the advice of an expert; redesigning is more expensive.
Finally, the annexes of the standard should be consulted. There are often very helpful guidelines or even tables that can be used as templates or checklists for compliance assessment.
Tip 4: A test plan needs a draft and a prototype needs pretests.
Essential Performance and risk analysis will have an impact on the testing requirements. For this reason, it is important to get these done before the test plan.
There are often many tests that are not applicable. Helbling’s experts recommend marking these as such and always explaining why they are not applicable to simplify future audits or reviews because what is not documented is not considered.
Early in the development cycle, an isolation diagram should be created and the voltage levels against which the device must be tested should be indicated. These diagrams are very easy to review with experts and help greatly in determining the test plan.
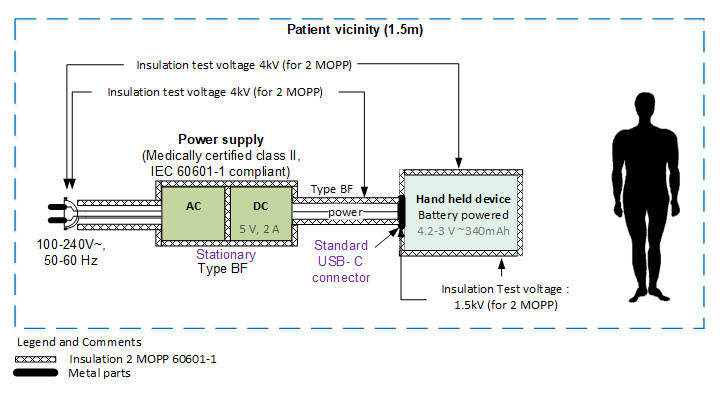
The standard requires performing certification tests with a representative sample of the device, but it is not ideal to go through all the effort of refining every detail of the design only to then find out that it requires redesigning because several tests have failed. Therefore, pretesting certain aspects related to the IEC 60601 in early prototypes is key.
Pretesting the device at one’s own facilities is especially helpful. In just a few hours, the chances of compliance with the standard can be assessed and different design solutions can be evaluated quickly.
Performing tests at an accredited test house is generally expensive and needs to be planned well in advance. Therefore, Helbling uses in-house facilities to perform ESD, EMC, and leakage current tests with calibrated equipment. Whenever a certificate or in-depth knowledge of one specific aspect of the standard is required, Helbling works with accredited test houses to complete testing.

Tip 5 – Consider the duration of the accreditation process in your plan.
The duration of the different parts of the accreditation process is very often underestimated. A common mistake is to reduce the total lead time to the duration of the tests only.
Test houses have limited calibrated resources (such as anechoic chambers). The availability of the test house should be checked and booked in advance.
The critical components of medical electrical equipment such as batteries or power adapters must be certified separately. In some cases, no certificate is available from the manufacturer and additional testing must be organized.
Compiling the risk management file is a lot of work. If it is done according to ISO 14971, most of the work is already done. However, this information must also be transferred or mapped into the test report form (CB scheme document) to demonstrate that the risk management file meets the requirements of the applicable IEC 60601 family of standards. Gathering all the information and completing this test report form can easily take a month or more.
Depending on the defined use environment and the presence of software, IEC 60601 certification may also require submission of the usability engineering file according to IEC 62366 and the software development file according to IEC 62304. This documentation must be ready in time.
Following document analysis, the test house may generate non-conformity reports requiring some adaptations or additions to the documentation. For example, warning information may need to be added to the instructions for use and safety signs on the product label.
After the tests and document analysis have been completed, a report is issued by the test house. Then, an independent reviewer examines the report to assess if the tests have been performed correctly and finally release the CB certificate for the device. This activity depends on the expert’s availability, so one to four weeks must be considered for the CB certificate release.
Summary: Integrating IEC 60601 standards into the design process is key to success
Considering the IEC 60601 family of standards early in the design process is challenging but crucial for the development of safe medical electrical equipment. Helbling has extensive experience in this area and can provide advice and assistance on product design, pretesting, and the certification process with its state-of-the-art infrastructure and equipment.
Authors: Laura Santos Carreras, Chantal Boyer Chardonnens, Stefan Bauer
Main Image: AdobeStock




